Definition
Phenylketonuria (PKU) is a rare condition in which a baby is born without the ability to properly break down an amino acid called phenylalanine.
Alternative Names
PKU; Neonatal phenylketonuria
Causes
PKU is inherited, which means it is passed down through families. Both parents must pass on a nonworking copy of the gene in order for a baby to have the condition. When this is the case, their children have a 1 in 4 chance of being affected.
Babies with PKU are missing an enzyme called phenylalanine hydroxylase. It is needed to break down the essential amino acid phenylalanine. Phenylalanine is found in foods that contain protein.
Without the enzyme, levels of phenylalanine build up in the body. This buildup can harm the central nervous system and cause brain damage.
Symptoms
Phenylalanine plays a role in the body's production of melanin. The pigment is responsible for skin and hair color. Therefore, infants with the condition often have lighter skin, hair, and eyes than brothers or sisters without the disease.
Other symptoms may include:
- Delayed mental and social skills
- Head size much smaller than normal
- Hyperactivity
- Jerking movements of the arms or legs
- Mental disability
- Seizures
- Skin rashes
- Tremors
If PKU is untreated, or if foods containing phenylalanine are eaten, the breath, skin, ear wax, and urine may have a "mousy" or "musty" odor. This odor is due to a buildup of phenylalanine substances in the body.
Exams and Tests
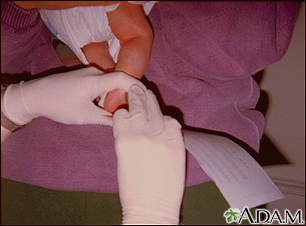
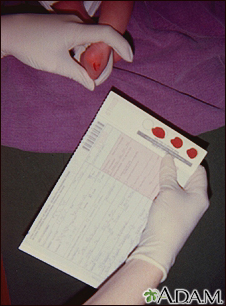
PKU can be easily detected with a simple blood test. All states in the United States require a PKU screening test for all newborns as part of the newborn screening panel. The test is generally done by taking a few drops of blood from the baby before the baby leaves the hospital.
If the screening test is positive, further blood and urine tests are required to confirm the diagnosis. Genetic testing is also done.
Treatment
PKU is a treatable disease. Treatment involves a diet that is very low in phenylalanine, particularly when the child is growing. The diet must be strictly followed. This requires close supervision by a registered dietitian or doctor, and cooperation of the parent and child. Those who continue the diet into adulthood have better physical and mental health than those who don't stay on it. "Diet for life" has become the standard most experts recommend. Women who have PKU need to follow the diet before conception and throughout pregnancy.
There are large amounts of phenylalanine in milk, eggs, and other common foods. The artificial sweetener NutraSweet (aspartame) also contains phenylalanine. Any products containing aspartame should be avoided.
There are several special formulas made for infants with PKU. These can be used as a protein source that is extremely low in phenylalanine and balanced for the remaining essential amino acids. Older children and adults use a different formula that provides protein in the amounts they need. People with PKU need to take formula every day for their entire life.
Outlook (Prognosis)
The outcome is expected to be very good if the diet is closely followed, starting shortly after the child's birth. If treatment is delayed or the condition remains untreated, brain damage will occur. School functioning may be mildly impaired.
If proteins containing phenylalanine are not avoided, PKU can lead to mental disability by the end of the first year of life.
Possible Complications
Severe mental disability occurs if the disorder is untreated. ADHD (attention-deficit hyperactivity disorder) appears to be a common problem in those who do not stick to a very low-phenylalanine diet.
When to Contact a Medical Professional
Call your health care provider if your infant has not been tested for PKU. This is particularly important if anyone in your family has the disorder.
Prevention
An enzyme assay or genetic testing can determine if parents carry the gene for PKU. Chorionic villus sampling or amniocentesis can be done during pregnancy to test the unborn baby for PKU.
It is very important that women with PKU closely follow a strict low-phenylalanine diet both before becoming pregnant and throughout the pregnancy. Buildup of phenylalanine will damage the developing baby, even if the child has not inherited the full disease.
References
Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Defects in metabolism of amino acids. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 103.
Kumar V, Abbas AK, Aster JC. Genetic and pediatric diseases. In: Kumar V, Abbas AK, Aster JC, eds. Robbins Basic Pathology. 10th ed. Philadelphia, PA: Elsevier; 2018:chap 7.
Vockley J, Andersson HC, Antshel KM, et al; American College of Medical Genetics and Genomics Therapeutics Committee. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188-200. PMID: 24385074 pubmed.ncbi.nlm.nih.gov/24385074/.