Definition
Gaucher disease is a rare genetic disorder in which a person lacks an enzyme called glucocerebrosidase (GBA).
Alternative Names
Glucocerebrosidase deficiency; Glucosylceramidase deficiency; Lysosomal storage disease - Gaucher
Causes
Gaucher disease is rare in the general population. People of Eastern and Central European (Ashkenazi) Jewish heritage are more likely to have this disease.
It is an autosomal recessive disease. This means that the mother and father must both pass one abnormal copy of the disease gene to their child in order for the child to develop the disease. A parent who carries an abnormal copy of the gene but doesn't have the disease is called a silent carrier.
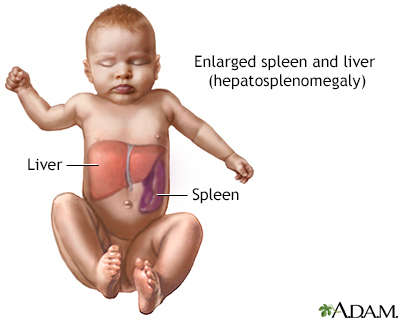
The lack of the GBA causes harmful substances to build up in the liver, spleen, bones, and bone marrow. These substances prevent cells and organs from working properly.
There are three main subtypes of Gaucher disease:
- Type 1 is most common. It involves bone disease, anemia, an enlarged spleen and low platelets (thrombocytopenia). Type 1 affects both children and adults. It is most common in the Ashkenazi Jewish population.
- Type 2 usually begins in infancy with severe neurologic involvement. This form can lead to rapid, early death.
- Type 3 may cause liver, spleen, and brain problems. People with this type may live into adulthood.
Symptoms
Bleeding because of low platelet count is the most common symptom seen in Gaucher disease. Other symptoms may include:
Exams and Tests
The health care provider will perform a physical exam and ask about the symptoms.
The following tests may be done:
Treatment
Gaucher disease can't be cured. But treatments can help control and may improve symptoms.
Medicines may be given to:
- Replace the missing GBA (enzyme replacement therapy) to help reduce spleen size, bone pain, and improve thrombocytopenia.
- Limit production of fatty chemicals that build up in the body.
Other treatments include:
- Medicines for pain
- Surgery for bone and joint problems, or to remove the spleen
- Blood transfusions
Support Groups
These groups can provide more information on Gaucher disease:
Outlook (Prognosis)
How well a person does depends on their subtype of the disease. The infantile form of Gaucher disease (Type 2) may lead to early death. Most affected children die before age 5.
Adults with the type 1 form of Gaucher disease can expect normal life expectancy with enzyme replacement therapy.
Possible Complications
Complications of Gaucher disease may include:
Prevention
Genetic counseling is recommended for prospective parents with a family history of Gaucher disease. Testing can determine if parents carry the gene that could pass on the Gaucher disease. A prenatal test can also tell if a baby in the womb has Gaucher syndrome.
References
Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Defects in metabolism of lipids. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 104.
Krasnewich DM, Sidransky E. Lysosomal storage diseases. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26th ed. Philadelphia, PA: Elsevier; 2020:chap 197.
Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18.